
Brain lobes
Your brain is made up of different sections which are responsible for different skills, thoughts, feelings, and….yes, sometimes; seizures. These sections are officially called “lobes” and they form the “anterior” aka the “outer” part of your brain known as the Cerebrum. Your brain has two halves known as hemispheres - the left hemisphere and the right hemisphere.
P.S. If you see a new word in the description of something, it should have its own description too, so look elsewhere on the page or in the general glossary!
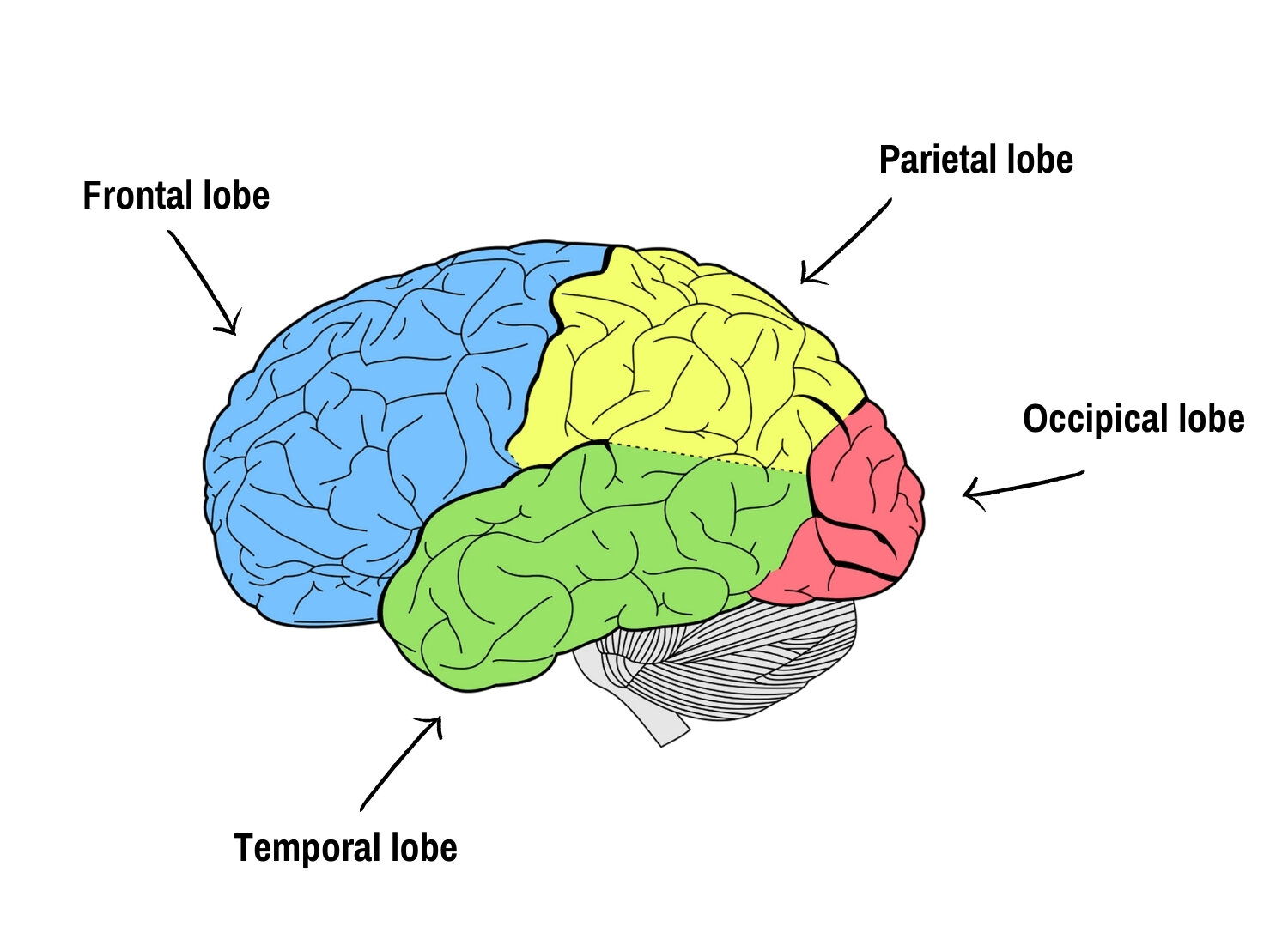
The lobes of the left hemisphere.
Join us on social
Cool facts
Fact: Your right hemisphere is responsible for movement on the left side of your body - and your left hemisphere (shown in the pictures) is responsible for movement on the right hand side of your body!
Fact: The brain is so, so, so complex that the best Neurologists and Neuroscientists in the world will openly tell you how little it is still understood and why we need more research to help people with epilepsy!
Fact: Lots of people have some brain damage and depending upon individual circumstances, other parts of the brain can take over the work .
Fact: Some children can have HALF of their brain removed (this is called a hemispherectomy ) as treatment for their epilepsy and still be quite a high functioning person - because the other half of their brain can take over!
Fact: Some people will have damage to their brain and develop epilepsy - but some people don’t.
Fact: Each half of your brain is connected by a tissue called the corpus callosum.
Fact: Each of your lobes connect and effectively communicate with each other. It’s like sharing data! When the information travels it’s called a neural pathway.
Fact: Some of the 86 billion neurons in your brain are tiny, and some are relatively long and spindly (we like the purkinje neuron (in your cerebellum -the grey bit in the picture of the brain) - check it out here)!
Fact: Each of your brain lobes tend to be divided into areas that serve very specific functions.
Fact: Each half of your brain looks very similar on the outside, but each generally have different “responsibilities”/functions. For example:
The left hemisphere of your brain is responsible for language and speech and is called the "dominant" hemisphere;
The right hemisphere of your brain plays a large part in interpreting visual information and spatial processing;
But…. (!) in about one third of people who are left-handed, speech function may be located on the right side of the brain!
Frontal lobe
Your Frontal lobe is (shockingly!) the front of your brain and right behind your forehead. It’s the largest lobe of your brain too.
It is involved in activities like muscle control, intellect, sequencing movements, problem solving, spontaneity, reasoning and judgement, personality, attention and concentration, mood, language, and social and sexual behaviour.

Parietal lobe
Your Parietal lobe is located behind your Frontal lobe - some might say in the middle-top of your head!
You use it in language and calculation, as well as in the perception of various sensations such as touch, pain, and pressure.
It enables you to interpret language, words, gives you a sense of touch, pain, temperature, interprets signals from vision, hearing, motor, sensory and memory, and gives you a spatial and visual perception.

Temporal lobe
Your Temporal lobe is located near your ear - next to both your Frontal and Parietal lobes.
It enables you to understand auditory information such as language and is responsible for memory, hearing, sequencing and organisation. This lobe is also believed to play an important role in processing emotions.
The dominant temporal lobe is the left side in most people and is involved in understanding language and learning and remembering verbal information. The non-dominant lobe, which is usually on the right hand side, is involved in learning and remembering non-verbal information (e.g. visuo-spatial material and music).
Temporal lobe epilepsy is the most common type of focal epilepsy.

Occipital lobe
Your Occipital lobe is at the back of your head, behind/next to your Parietal and your Temporal lobe.
It is involved in processing visual stimuli; enabling you to interpret colour, light, and movement.
The Occipital lobe takes information from the retina in the eye and translates it into the visual world.

If you have focal seizures in one lobe of your brain (which could be the Frontal, Temporal, Occipital or Parietal lobe), then the function of that lobe will be disrupted. But, remember, because that lobe is connected to the rest of your brain too, functions there can also be disrupted.
Of course, if you have focal to bilateral tonic-clonic or generalised seizures, then there’s no messing around because all lobes will be affected during the generalised phase.
- ALDH7A1
- Adeno-Associated Virus
- Angelman Syndrome
- Anxiety
- Art
- Australia
- Awareness
- Brain scan
- CDKL5
- Cambridge
- Cerebral palsy
- Cerrubellum
- Childhood epilepsy
- Culture
- Depression
- Diagnosis
- Discrimination
- Dissociative Seizures
- Doose Syndrome
- Dravet
- EEG
- Education
- Employment
- EpSMon
- Epilepsy Nurse
- Family
- GLUT1
- Gelastic
- Genetic Epilepsy Team Australia
- Genetics
- Genome Scientist
- Global Genes
- Hope for Hypothalamic Hamartomas
- Hypothalamic Harmartoma
- India
- Infantile Spasms
- Injury
- Ireland
- KCNQ2
- Men
- Mental Health
- Music
- Netherlands
- NeuroRenee
- Neurophysiology
- Neuroscience
- Neuroscientist
- NightWatch
- Non-Epileptic Seizure
- PNES